
Conotruncal anomalies raise an immediate set of challenges: estimating genetic risk, selecting the right test, and counselling families with precision. Most of these defects are surgically correctable, but their strong association with 22q11.2 deletion syndrome (DiGeorge syndrome) often defines the long-term outcome and counselling complexity more than the cardiac lesion itself.
This blog provides a systematic methodology for integrating genetic assessment into fetal cardiac evaluation, offering evidence-based protocols for risk stratification, testing strategies, and family communication.
Conotruncal Anomalies
A conotruncal anomaly refers to a spectrum of congenital heart defects arising from abnormal septation or rotation of the embryonic conotruncus, the region formed by the conus (infundibulum) and truncus arteriosus, that gives rise to the ventricular outflow tracts, semilunar valves, and great arteries.

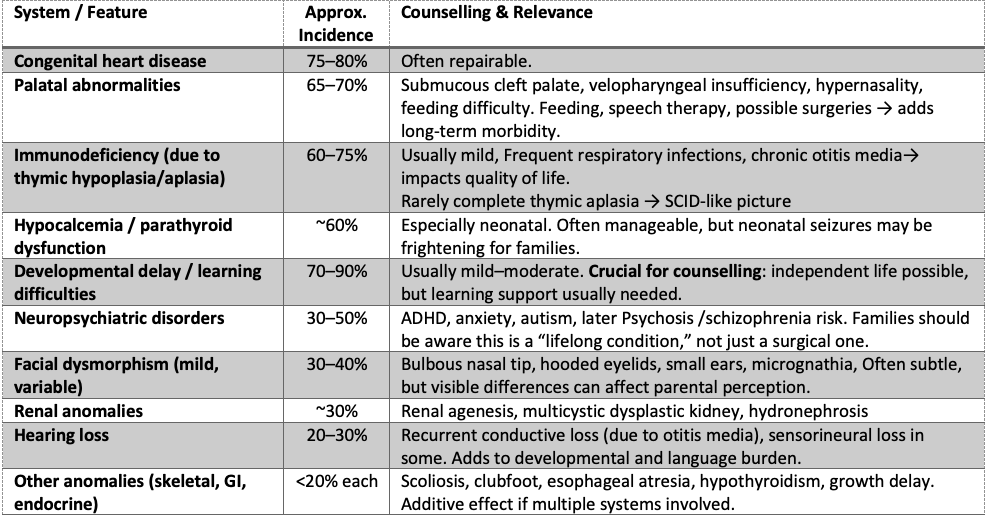
Common Manifestations of 22q11.2 Deletion Syndrome (DiGeorge Syndrome)
Methodology Framework: The 5-Step Genetic Assessment Protocol
Step 1: Comprehensive Phenotypic Characterisation
Cardiac Anatomy Documentation
– Perform detailed segmental analysis of all cardiac structures
– Document specific Conotruncal defect type and severity
– Assess aortic arch sidedness and branching patterns
– Evaluate pulmonary artery anatomy and valve morphology
Thymic Assessment Protocol
– Obtain three-vessel view at the level of Trachea (3VT)
– Calculate the thymic-thoracic ratio using a standardised technique
– Normal ratio: 0.44 ± 0.05
– Suspicious ratio: <0.25 (90% sensitivity, 98.5% specificity for 22q11.2 deletion) – Document thymic presence, size, and echogenicity

Systematic Extracardiac Survey
– CNS: Cavum septum pellucidum dimensions, ventricular asymmetry
– Craniofacial: Ear length percentiles, facial profile assessment
– Musculoskeletal: Vertebral alignment, limb measurements
– Genitourinary: Renal size, pelviectasis
– Amniotic fluid: Quantitative assessment of volume
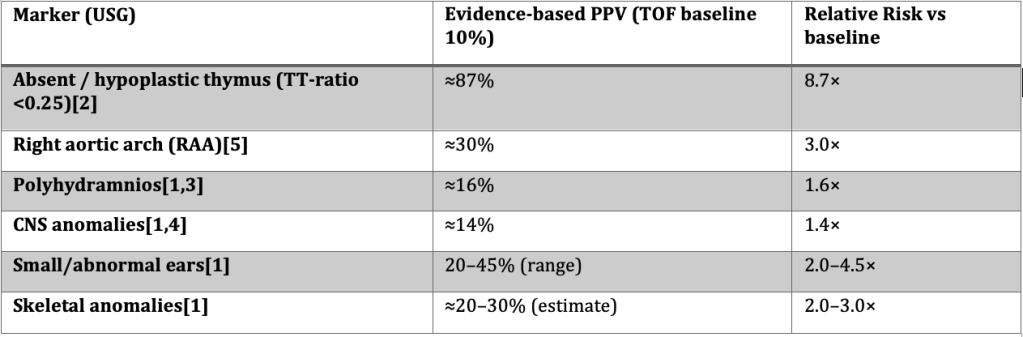
1.Schindewolf E, et al.; 2. Chaoui R et al. ; 3. Simonyi A, et al. ; 4. Khalil A, et al.; 5. Goldmuntz E, et al.
Examples
Example 1 — Absent thymus:
Baseline risk (TOF) = 10%. Using Chaoui’s sensitivity (90%) and specificity (98.5%), Bayes calculation → PPV ≈ 87%. Interpretation: almost 9× baseline.
Example 2 — Polyhydramnios + CNS anomaly:
Baseline risk (TOF) = 10%. Schindewolf frequencies: polyhydramnios 31%, CNS 38%. Using CHD prevalence data (Simonyi, Khalil) → combined PPV ≈ 21%. Interpretation: doubles baseline, but not as strong as the thymus finding.

Step 2: Evidence-Based Risk Stratification
This updated stratification reflects recalculated relative risks and published evidence. It highlights that some extracardiac markers (e.g., polyhydramnios, CNS anomalies) increase risk modestly and should be seen as risk modifiers, not standalone high-risk features.
High-Risk Group (Genetic yield >40%)
– Interrupted aortic arch Type B (baseline risk ≈50%)
– Any conotruncal defect + thymic hypoplasia/aplasia (PPV ≈87%, RR ≈9×)
– Non-isolated conotruncal anomalies with ≥2 extracardiac systems involved
Moderate-Risk Group (Genetic yield ~20–40%)
– Truncus arteriosus (isolated yield ≈30–35%)
– Tetralogy of Fallot + right aortic arch (≈25–30%)
– TOF with multiple “soft” extracardiac markers (e.g., polyhydramnios + CNS, or ear + skeletal anomalies) → combined PPV ~20–25%
Lower-Risk Group (Genetic yield <20%)
– Isolated Tetralogy of Fallot without extracardiac markers (≈6–10%)
– TOF with a single modest marker only (polyhydramnios OR CNS anomaly alone → ≈14–16%)
– Other conotruncal variants without extracardiac features
Counselling tip: Thymus hypoplasia and IAA-B remain the strongest predictors. Polyhydramnios and CNS anomalies are best understood as modifiers that modestly raise pre-test probability. The presence of multiple extracardiac markers shifts a case from baseline into the moderate-yield group.
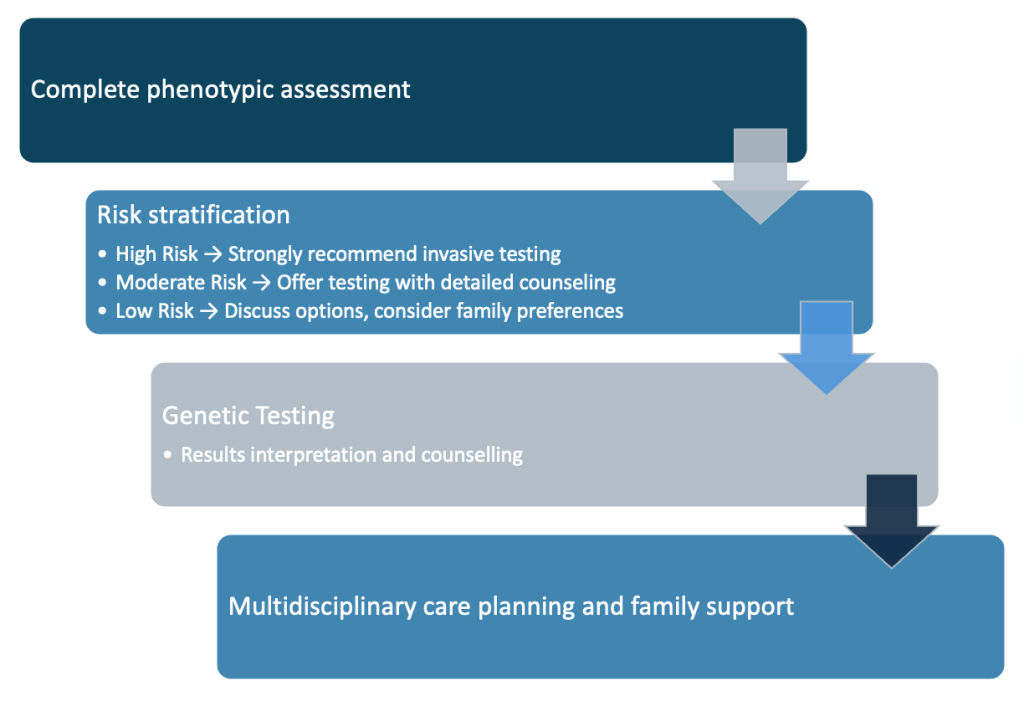
Step 3: Genetic Testing Algorithm
Primary Testing Strategy
– First-line: Chromosomal microarray analysis (CMA)
– Detects aneuploidies and pathogenic CNVs
– Recommended for all moderate and high-risk cases
– Turnaround time: 10-14 days
Secondary Testing Considerations
– Whole exome sequencing (WES): For CMA-negative cases with high clinical suspicion
– Rapid FISH testing: For 22q11.2 deletion if urgent results needed
– Targeted gene panels: For specific syndromic presentations
Testing Timing Methodology
– CVS: 11-13 weeks (earlier genetic information)
– Amniocentesis: 15-20 weeks (lower procedure risk)
– Consider maternal preference and procedural expertise
Step 4: Structured Counselling Approach
Pre-test Counselling Framework
- Risk communication: discuss possibilities and percentages
- Amniocentesis Procedure risks: the risk of miscarriage is only 0.2-0.3%.
- Result implications: Explain possible outcomes and next steps
- Family values assessment: Understand decision-making preferences
Post-test Result Communication
- Positive results: Immediate genetics referral, multidisciplinary planning
- Negative results: Discuss residual risk, ongoing monitoring
- Variants of uncertain significance: Explain limitations and follow-up
| “No two children are exactly alike. We can’t predict everything, but we do know that early detection and coordinated care can make a huge difference in your baby’s quality of life and that you will have a dedicated team by your side, every step of the way. This is a big journey, and we’ll support you whatever path you choose.” |
Step 5: Multidisciplinary Care Coordination
Multidisciplinary Team
– Maternal-fetal medicine specialist
– Clinical geneticist/genetic counsellor
– Pediatric cardiologist
– Neonatologist
– Social worker (family support)
Care Transition Planning
– Delivery centre selection (tertiary care with cardiac surgery)
– Postnatal evaluation protocol
– Early intervention services coordination
– Family support resources
Clinical Decision-Making Algorithm
Conclusion
Early, detailed diagnosis, paired with thoughtful and empathetic counselling, and risk-based genetic testing, empowers families. It transforms prenatal fear into a proactive roadmap, allowing every child, no matter the diagnosis, the best chance for a healthy and meaningful life.
1. Not all CHDs carry equal genetic risk—phenotype matters.
2. Thymic evaluation is essential in conotruncal anomalies.
3. Prioritise genetic testing for high-risk structural patterns or non-isolated defects.
4. Counselling should balance medical facts with emotional support and family values.
5. Early, coordinated care planning improves outcomes and parental preparedness.
References and suggested reading.
- McDonald-McGinn DM, et al. 22q11.2 Deletion Syndrome. GeneReviews, NCBI Bookshelf. (Updated online review; authoritative clinical summary for phenotype, genetics, and counselling).
- Chaoui R, Heling KS, Lopez AS, Thiel G, Karl K. The thymic-thoracic ratio in fetal heart defects: a simple way to identify fetuses at high risk for microdeletion 22q11. Ultrasound Obstet Gynecol. 2011;37(4):397–403. DOI:10.1002/uog.8952. (Key paper on the TT-ratio — practical screening tool for fetal imaging teams).
- Goldmuntz E, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32(2):492–498. (Seminal paper reporting deletion rates by lesion — e.g., IAA, truncus, TOF).
- Schindewolf E, Khalek N, Johnson MP, et al. Expanding the fetal phenotype: prenatal sonographic findings and perinatal outcomes in a cohort of patients with a confirmed 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(8):1735–1741.(Good prenatal cohort describing extracardiac features and outcomes; useful for counselling).
- Simonyi A, Eros FR, Hajdu J, Beke A. Effectiveness of fetal ultrasound diagnostics in cardiac malformations and association with polyhydramnios and oligohydramnios. Quant Imaging Med Surg. 2021 Jul;11(7):2994-3004. doi: 10.21037/qims-20-823.
- Óskarsdóttir S, et al. Updated clinical practice recommendations for managing children and adolescents with 22q11.2 deletion syndrome (22q11.2DS). (Recent expert consensus/guideline update, practical management & follow-up recommendations). 2022/2023. (Use for counselling on multisystem surveillance and long-term care).
